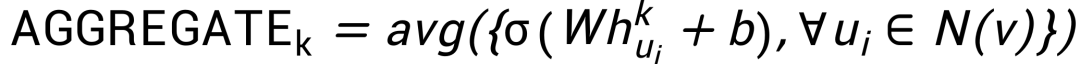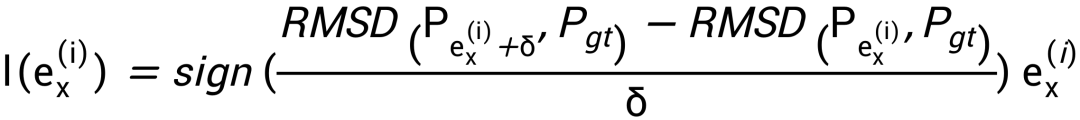由百度飞桨举办的螺旋桨RNA结构预测竞赛圆满结束。本次竞赛依托百度研究院在RNA结构预测上的算法优势和百度大脑AI Studio平台优势,汇聚生物计算、人工智能领域从业者和爱好者,探索人工智能技术在生物计算上的技术应用。本次比赛邀请北京大学生命科学学院刘君教授团队提供实验数据,构建测试集,保证比赛的科学性和公正性。各获胜队伍已在比赛页揭晓。
上周我们解锁了冠军团队的技术方案,今天又有幸邀请到亚军和季军队伍分享各自的技术成果。话不多说,干货马上呈现!
亚军-FAAR团队
成员:朱强(南京大学博士后)、胡礼沐(南京大学博士)、顾旭(南京大学研究生)、崔子宜(UCLA本科生)和周远哲(数据挖掘爱好者)。周、顾、崔和胡同学负责模型搭建与调试;朱、胡同学负责分析预测结果并提出可能的改进方向。
季军-FIT团队
成员:许陈成(清华大学博士)、周建宇(博士、南开大学讲师)、刘桥(斯坦福大学博士后)。许同学主要研究生物信息领域的深度学习算法设计,周同学主要研究RNA可变剪接分析与二级结构预测算法设计,刘同学主要研究人工智能算法在临床数据、测序数据、药理学数据的分析和应用。
赛题及算法介绍
RNA是本次螺旋桨结构预测比赛的主角。比赛要求用深度学习的方法预测每个碱基的不配对概率。作为一个线性的链状大分子,RNA具有非常高的柔性,易于弯曲,好比一条细长的线。如果碱基之间没有任何相互作用,RNA在三维空间里就会杂乱无章,毫无结构而言。可作为生物大分子的RNA需要具有特定结构才能行使其生物功能, 因而碱基间是否配对和如何配对至关重要。
百度研究院近年来在RNA结构预测上做出了重要贡献, 研发出一系列速度最快同时准确度极高的线性算法, 做到了对RNA最小自由能(MFE)结构的高效预测(LinearFold), 对碱基配分和配对概率的高效预测(LinearPartition), 对RNA序列的设计优化(LinearDesign), 和对同源RNA序列的结构分析(LinearTurboFold)。这次比赛主要是基于百度研究院LinearFold和LinearPartition算法,用深度学习方法预测RNA的不配对概率。
RNA碱基不配对概率可以用在疫苗和药物设计上。RNA病毒是以RNA为遗传物质的病毒,我们熟知的新冠、HIV、流感病毒等都属于RNA病毒。病毒RNA的结构对其生理功能有重要影响,如最近发表的一些研究就通过分析新冠病毒的二级结构寻找药物靶点。
同时,新冠病毒具有已知最长的RNA基因组(30,000碱基左右),如何在基因组全长尺度上分析其二级结构,仍然很有挑战。另一方面,RNA碱基不配对概率可以用在疫苗序列设计上。拿mRNA疫苗为例, 如果mRNA序列里大部分碱基有很小的不配对概率, 说明这是一个稳定的二维甚至三维RNA结构。稳定的结构会大大延长其在溶液和细胞内的寿命,从而提高蛋白的产量。
反之,如果大部分碱基有非常大的不配对概率, 说明这是一个非常无结构或动态的RNA,会让mRNA在溶液中易于被水解, 降低其稳定性和寿命, 不利于运输、储藏、翻译等。所以碱基不配对概率对RNA序列设计有重要的定量指导意义。
亚军团队方案详解
1. 赛题及难点分析
RNA的二级结构即2D平面上的碱基配对结构,可以用点括号表示法体现:使用点“.”和成对的括号“(”和“)”组成的序列表示该二级结构。预测碱基不配对概率可以理解为:预测RNA序列中各碱基对应位置上的二级结构是“.”的概率。预测结果是一个一维的由范围在0~1之间数字组成的序列。可见,这是深度学习中典型的N-N的seq2seq问题。
本赛题的两个难点如下:
(1)用深度学习模型预测不配对概率的相关文献较少,更多的文献是拿不配对概率这一关键性质去预测其他的相关性质,这意味着需要基于对生化知识和深度学习模型的理解去发挥想象并不断试错;
(2)本次训练集相对较小,而预测集相对训练集又有一定的差异,这要求预测模型更robust,增加了赛题的难度。
2. 模型搭建思路
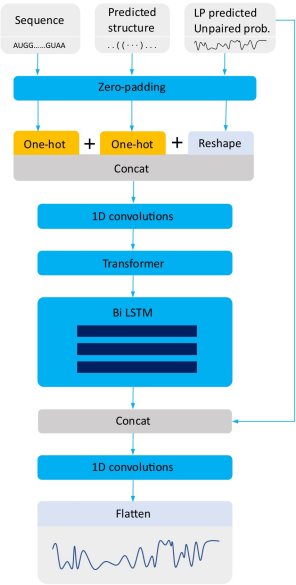
本赛题类似于机器翻译任务,特点是词汇表很小,但序列很长。因此初期搭建模型时,首先使用自然语言处理中常用的词向量嵌入(Word Embedding)方法,将每个位置的碱基转化为向量;然后进行特征提取,使信息更加集中;最后使用循环神经网络或者其他处理序列信息方法,学习序列不同位置的依赖关系。搭建模型时用到的深度学习方法的关键代码以及模型结构图如下:
首先需要解决的是输入序列的编码问题。分别尝试了word-embedding和one-hot方法后,发现one-hot编码得到的结果要好于word-embedding。在NLP任务中,字词编码的词表非常大,导致多维且稀疏,所以需要word-embedding得到词语的低维稠密表示。这样做的好处是不同字词间有很大的联系,而这一联系可以通过词向量间的cos距离等来刻画。
而RNA碱基种类只有4种,只需要一个4维的one-hot向量就可以表示。此外,不同碱基间的关联度小,差异性才是更重要的特点。one-hot向量可以充分表达差异性,因为不同one-hot向量在高维空间中相互垂直。此外,使用paddlehelix的linear_rna预测的概率与one-hot拼接,作为输入,具体可参考:
https://github.com/PaddlePaddle/PaddleHelix
K-mers with 1D convolutions
NLP任务中,有时会用到n-gram技术,即将多个词绑定为一个整体。这个技术在蛋白质序列、DNA序列以及基因组相关的研究中也经常使用,称为k-mers[1]。我们将一维卷积和k-mers结合起来,通过CNN捕获局部信息,使其具有生物学意义。使用k-mers构建词表时,假设k=5,那么词表的大小就是4^5=1024。这种处理相当于在编码时将一定范围内的上下文也考虑进去,增加了词的多样性,可以在一定程度上提高模型的学习能力。经过调试,k-mers长度为9,即使用9-mer时,预测效果最好。
Transformer中multi-head允许不同的头学习输入不同的隐藏层表示,可以提高预测性能。此外,self-attention机制允许我们先前构造的每个k-mer注意到所有的k-mer,很好地解决了序列内长距离的依赖问题。Transformer block的paddle代码实现如下:
class TransformerBlock(Layer):
def __init__(self, embed_dim, num_heads, ff_dim, rate=0.1):
super(TransformerBlock, self).__init__()
self.att = MultiHeadAttention(embed_dim=embed_dim, num_heads=num_heads)
self.fc1 = Linear(embed_dim, ff_dim)
self.act1 = ReLU()
self.fc2 = Linear(ff_dim, embed_dim)
self.layernorm1 = LayerNorm(normalized_shape=(embed_dim), epsilon=1e-6)
self.layernorm2 = LayerNorm(normalized_shape=(embed_dim), epsilon=1e-6)
self.dropout1 = Dropout(rate)
self.dropout2 = Dropout(rate)
def forward(self, inputs):
attn_output = self.att(inputs, inputs)
attn_output = self.dropout1(attn_output)
out1 = self.layernorm1(inputs + attn_output)
ffn_output = self.fc1(out1)
ffn_output = self.act1(ffn_output)
ffn_output = self.fc2(ffn_output)
# ffn_output = self.ffn(out1)
ffn_output = self.dropout2(ffn_output)
return self.layernorm2(out1 + ffn_output)
尝试仅使用transformer、仅使用Bi LSTM和同时使用transformer和Bi LSTM后,结果发现同时使用transformer和bi LSTM,且bi LSTM层数为3时,预测效果最好。
3. 模型结果分析
本方案主要尝试了三个改进方向:
词向量嵌入:Bert预训练、 Embedding(paddle.fluid)、 one_hot;
序列特征学习:序列卷积(残差结构);
类机器翻译(计算每个位置的ACGU是点的概率,由于类似机器翻译,故称之为类机器翻译):LSTM、Transformer。
基于以上三个改进方向,通过组合模型,得到三种模型在A榜数据的表现:
Model1 |
Model2 |
Model3 |
|
RMSD on B board |
0.246 |
0.243 |
0.241 |
* 交叉验证后,model3的最好成绩为0.239
4. 总结分析
使用Bert作预训练没有起到太好的效果,未来需要在这方面做更深入的研究;此外,为了发挥模型的科研价值,需使用更大的数据集更深层地研究其化学含义及解释;再次,从这次比赛中也收获了很多经验, 今后将考虑进一步用深度学习和机器学习方法预测DNA、RNA和各种分子及材料的化学性质。
GitHub源代码:
https://github.com/ziyicui2022/FAAR-ML.git
季军团队方案详解
1. 方案介绍
整体方案如下图所示,考虑到数据中同时有序列信息和图信息,分别使用双向LSTM提取序列信息,用图卷积神经网络GCN提取拓扑结构信息。由于训练数据较少,为降低过拟合的影响,先结合经典热力学方法和深度学习方法预测经典动力学方法中的能量参数(stacking energy),再使用ViennaRNA中提供的动态规划过程推测在给定能量下序列的不配对概率。
2. 模型搭建
首先,在飞桨中实现graphsage[2]。Graphsage包括聚合(aggregation)和更新两个阶段,聚合阶段使用平均池化,即:
其中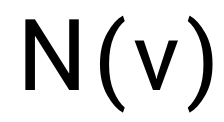

class egsage(Layer):
def __init__(self, in_channels, out_channels, edge_channels):
super(egsage, self).__init__()
self.in_channels=in_channels
self.out_channels=out_channels
self.edge_channels=edge_channels
self.message_lin=partial(paddle.fluid.layers.fc, size=out_channels, act="relu")
self.agg_lin=partial(paddle.fluid.layers.fc, size=out_channels, act="relu")
def forward(self, x, edge_attr, edge_index):
m_j=fluid.layers.gather(x, edge_index[1])
m_j=fluid.layers.reshape(paddle.fluid.layers.concat(input=[m_j, edge_attr], axis=-1), [-1, 1024+1])
m_j=self.message_lin(m_j)
#aggr
aggr_out=fluid.layers.scatter(x*0.0, edge_index[0], m_j)
aggr_size=fluid.layers.scatter(x*0.0, edge_index[0], m_j*0.0+1.0)
aggr_out=aggr_out/(aggr_size+1e-9)
#update
aggr_out=fluid.layers.concat(input=[x, aggr_out], axis=1)
aggr_out=self.agg_lin(aggr_out)
return aggr_out
然后,构建完整的神经网络模型。先将序列碱基和LinearFold预测结构中的点与括号嵌入为向量,再传入双向LSTM和GCN中提取信息,代码如下所示。
class Model(Layer):
def __init__(self, lr):
super(Model, self).__init__()
#Embedding
self.seq_embedding = partial(paddle.fluid.embedding, size=(4, 128), is_sparse=False)
self.bra_embedding=partial(paddle.fluid.embedding, size=(3, 128), is_sparse=False)
#Bidirectional LSTM
self.net1=partial(paddle.fluid.layers.dynamic_lstm, size=128*16, use_peepholes=False)
self.net2=partial(paddle.fluid.layers.dynamic_lstm, size=128*16, use_peepholes=False, is_reverse=True)
#GCN
self.gcn1=egsage(256*4, 256*4, 1)
self.gcn2=egsage(256*4, 256*4, 1)
self.gcn3=egsage(256*4, 256*4, 1)
#FCs
self.inp_fc=partial(paddle.fluid.layers.fc, size=128, act="relu")
self.seq_fc=partial(paddle.fluid.layers.fc, size=128, act="relu")
self.fc=partial(paddle.fluid.layers.fc, size=128*16, act="relu")
def forward(self, seq, dot, edge, edge_attr, fgt):
# Embed
emb_seq=self.seq_embedding(seq)
emb_dot=(self.bra_embedding(dot))
emb_dot=self.inp_fc(emb_dot)
emb_seq=self.seq_fc(emb_seq)
emb=self.fc(paddle.fluid.layers.concat(input=[emb_seq, emb_dot], axis=1))
# Bidirectional LSTM
emb1, _=self.net1(emb)
emb2, _=self.net2(emb)
emb=paddle.fluid.layers.concat(input=[emb1, emb2], axis=1)
# GraphSage
emb_g=self.gcn1(emb, edge_attr, edge)
emb_g = self.gcn2(emb_g, edge_attr, edge)
emb_g = self.gcn3(emb_g, edge_attr, edge)
# Final embedding
emb=paddle.fluid.layers.concat([emb, emb_g], -1)
3. 模型优化
由于A榜和B榜测试集分布差异较大,我们在A榜和B榜采用不同的模型优化策略。A榜数据集,由于RNA长度较长,动态规划耗时较长,可直接使用神经网络预测不配对概率,损失函数为预测值和基准值之间的RMSD。B榜数据集,可结合经典的动力学方法和深度学习方法,对RNA序列得到神经网络预测的一组能量参数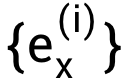
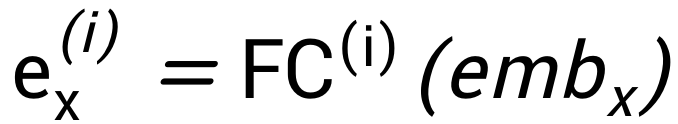
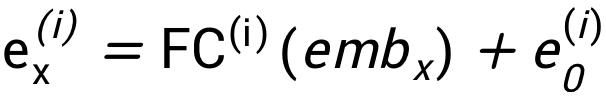
训练优化目标为降低的碱基不配对概率预测值和基准值之间的RMSD,主要难点在于动态规划过程不可导,不能直接使用梯度回传。为解决参数优化问题,参考强化学习训练技巧,对每个能量参数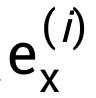
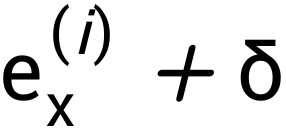
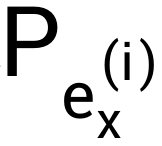
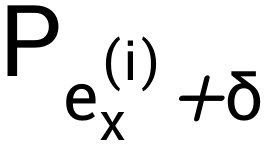
其中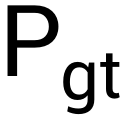
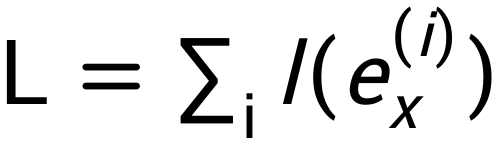
4. 结果分析
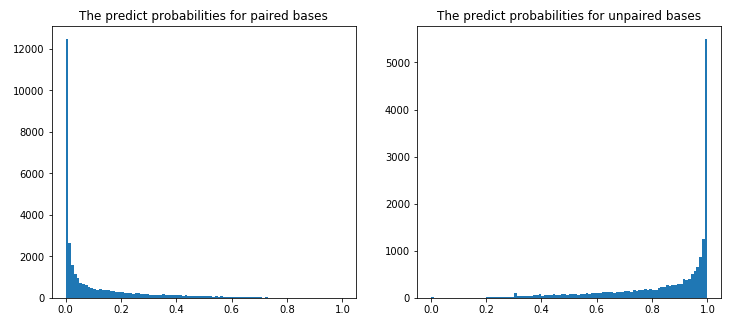
优化后模型的预测结果表明,对LinearFold预测结构中配对的位点,本方案预测的不配对概率绝大多数位于0附近(左图);反之,对LinearFold预测结构中不配对的位点,本方案预测的不配对概率绝大多数位于1附近(右图),与直觉一致。
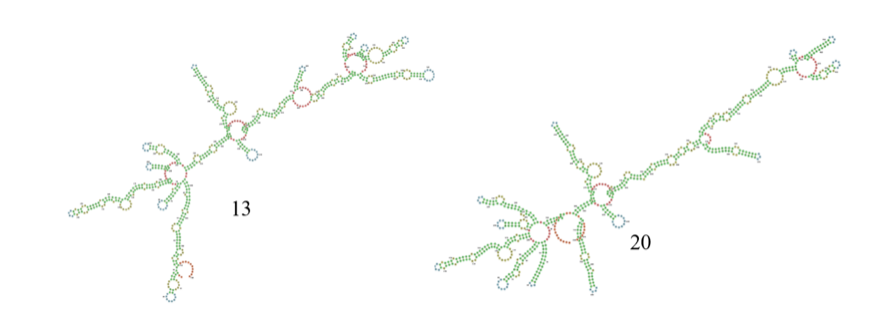
在比赛过程中,少数较长的、预测结果与LinearFold预测的配对情况相差较大的序列对最终成绩的影响较大,如第13条RNA序列和第20条RNA序列。为进一步提高分数,我们将LinearFold给出的结构转化为二值的不配对概率,然后比较模型预测的不配对概率P与

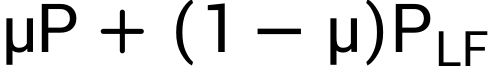
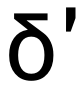
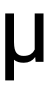
最终在结合了双向LSTM、GCN、强化学习训练技巧和对预测值的提炼后,本方案在A榜阶段获第一名,B榜阶段获第三名。
GitHub源代码:
https://github.com/Zoesgithub/Paddlepaddle-RNA-structure-prediction-3rd-place-.git
往期回顾:

参考文献
如在使用过程中有问题,可加入官方QQ群进行交流:793866180。
如果您想详细了解更多飞桨的相关内容,请参阅以下文档。
·飞桨官网地址·
https://www.paddlepaddle.org.cn/
·飞桨开源框架项目地址·
GitHub: https://github.com/PaddlePaddle/Paddle
Gitee: https://gitee.com/paddlepaddle/Paddle
长按上方二维码立即star!
飞桨(PaddlePaddle)以百度多年的深度学习技术研究和业务应用为基础,集深度学习核心训练和推理框架、基础模型库、端到端开发套件和丰富的工具组件于一体,是中国首个自主研发、功能丰富、开源开放的产业级深度学习平台。飞桨企业版针对企业级需求增强了相应特性,包含零门槛AI开发平台EasyDL和全功能AI开发平台BML。EasyDL主要面向中小企业,提供零门槛、预置丰富网络和模型、便捷高效的开发平台;BML是为大型企业提供的功能全面、可灵活定制和被深度集成的开发平台。
END

由百度飞桨举办的螺旋桨RNA结构预测竞赛圆满结束。本次竞赛依托百度研究院在RNA结构预测上的算法优势和百度大脑AI Studio平台优势,汇聚生物计算、人工智能领域从业者和爱好者,探索人工智能技术在生物计算上的技术应用。本次比赛邀请北京大学生命科学学院刘君教授团队提供实验数据,构建测试集,保证比赛的科学性和公正性。各获胜队伍已在比赛页揭晓。
上周我们解锁了冠军团队的技术方案,今天又有幸邀请到亚军和季军队伍分享各自的技术成果。话不多说,干货马上呈现!
亚军-FAAR团队
成员:朱强(南京大学博士后)、胡礼沐(南京大学博士)、顾旭(南京大学研究生)、崔子宜(UCLA本科生)和周远哲(数据挖掘爱好者)。周、顾、崔和胡同学负责模型搭建与调试;朱、胡同学负责分析预测结果并提出可能的改进方向。
季军-FIT团队
成员:许陈成(清华大学博士)、周建宇(博士、南开大学讲师)、刘桥(斯坦福大学博士后)。许同学主要研究生物信息领域的深度学习算法设计,周同学主要研究RNA可变剪接分析与二级结构预测算法设计,刘同学主要研究人工智能算法在临床数据、测序数据、药理学数据的分析和应用。
赛题及算法介绍
RNA是本次螺旋桨结构预测比赛的主角。比赛要求用深度学习的方法预测每个碱基的不配对概率。作为一个线性的链状大分子,RNA具有非常高的柔性,易于弯曲,好比一条细长的线。如果碱基之间没有任何相互作用,RNA在三维空间里就会杂乱无章,毫无结构而言。可作为生物大分子的RNA需要具有特定结构才能行使其生物功能, 因而碱基间是否配对和如何配对至关重要。
百度研究院近年来在RNA结构预测上做出了重要贡献, 研发出一系列速度最快同时准确度极高的线性算法, 做到了对RNA最小自由能(MFE)结构的高效预测(LinearFold), 对碱基配分和配对概率的高效预测(LinearPartition), 对RNA序列的设计优化(LinearDesign), 和对同源RNA序列的结构分析(LinearTurboFold)。这次比赛主要是基于百度研究院LinearFold和LinearPartition算法,用深度学习方法预测RNA的不配对概率。
RNA碱基不配对概率可以用在疫苗和药物设计上。RNA病毒是以RNA为遗传物质的病毒,我们熟知的新冠、HIV、流感病毒等都属于RNA病毒。病毒RNA的结构对其生理功能有重要影响,如最近发表的一些研究就通过分析新冠病毒的二级结构寻找药物靶点。
同时,新冠病毒具有已知最长的RNA基因组(30,000碱基左右),如何在基因组全长尺度上分析其二级结构,仍然很有挑战。另一方面,RNA碱基不配对概率可以用在疫苗序列设计上。拿mRNA疫苗为例, 如果mRNA序列里大部分碱基有很小的不配对概率, 说明这是一个稳定的二维甚至三维RNA结构。稳定的结构会大大延长其在溶液和细胞内的寿命,从而提高蛋白的产量。
反之,如果大部分碱基有非常大的不配对概率, 说明这是一个非常无结构或动态的RNA,会让mRNA在溶液中易于被水解, 降低其稳定性和寿命, 不利于运输、储藏、翻译等。所以碱基不配对概率对RNA序列设计有重要的定量指导意义。
亚军团队方案详解
1. 赛题及难点分析
RNA的二级结构即2D平面上的碱基配对结构,可以用点括号表示法体现:使用点“.”和成对的括号“(”和“)”组成的序列表示该二级结构。预测碱基不配对概率可以理解为:预测RNA序列中各碱基对应位置上的二级结构是“.”的概率。预测结果是一个一维的由范围在0~1之间数字组成的序列。可见,这是深度学习中典型的N-N的seq2seq问题。
本赛题的两个难点如下:
(1)用深度学习模型预测不配对概率的相关文献较少,更多的文献是拿不配对概率这一关键性质去预测其他的相关性质,这意味着需要基于对生化知识和深度学习模型的理解去发挥想象并不断试错;
(2)本次训练集相对较小,而预测集相对训练集又有一定的差异,这要求预测模型更robust,增加了赛题的难度。
2. 模型搭建思路
本赛题类似于机器翻译任务,特点是词汇表很小,但序列很长。因此初期搭建模型时,首先使用自然语言处理中常用的词向量嵌入(Word Embedding)方法,将每个位置的碱基转化为向量;然后进行特征提取,使信息更加集中;最后使用循环神经网络或者其他处理序列信息方法,学习序列不同位置的依赖关系。搭建模型时用到的深度学习方法的关键代码以及模型结构图如下:
首先需要解决的是输入序列的编码问题。分别尝试了word-embedding和one-hot方法后,发现one-hot编码得到的结果要好于word-embedding。在NLP任务中,字词编码的词表非常大,导致多维且稀疏,所以需要word-embedding得到词语的低维稠密表示。这样做的好处是不同字词间有很大的联系,而这一联系可以通过词向量间的cos距离等来刻画。
而RNA碱基种类只有4种,只需要一个4维的one-hot向量就可以表示。此外,不同碱基间的关联度小,差异性才是更重要的特点。one-hot向量可以充分表达差异性,因为不同one-hot向量在高维空间中相互垂直。此外,使用paddlehelix的linear_rna预测的概率与one-hot拼接,作为输入,具体可参考:
https://github.com/PaddlePaddle/PaddleHelix
K-mers with 1D convolutions
NLP任务中,有时会用到n-gram技术,即将多个词绑定为一个整体。这个技术在蛋白质序列、DNA序列以及基因组相关的研究中也经常使用,称为k-mers[1]。我们将一维卷积和k-mers结合起来,通过CNN捕获局部信息,使其具有生物学意义。使用k-mers构建词表时,假设k=5,那么词表的大小就是4^5=1024。这种处理相当于在编码时将一定范围内的上下文也考虑进去,增加了词的多样性,可以在一定程度上提高模型的学习能力。经过调试,k-mers长度为9,即使用9-mer时,预测效果最好。
Transformer中multi-head允许不同的头学习输入不同的隐藏层表示,可以提高预测性能。此外,self-attention机制允许我们先前构造的每个k-mer注意到所有的k-mer,很好地解决了序列内长距离的依赖问题。Transformer block的paddle代码实现如下:
class TransformerBlock(Layer):
def __init__(self, embed_dim, num_heads, ff_dim, rate=0.1):
super(TransformerBlock, self).__init__()
self.att = MultiHeadAttention(embed_dim=embed_dim, num_heads=num_heads)
self.fc1 = Linear(embed_dim, ff_dim)
self.act1 = ReLU()
self.fc2 = Linear(ff_dim, embed_dim)
self.layernorm1 = LayerNorm(normalized_shape=(embed_dim), epsilon=1e-6)
self.layernorm2 = LayerNorm(normalized_shape=(embed_dim), epsilon=1e-6)
self.dropout1 = Dropout(rate)
self.dropout2 = Dropout(rate)
def forward(self, inputs):
attn_output = self.att(inputs, inputs)
attn_output = self.dropout1(attn_output)
out1 = self.layernorm1(inputs + attn_output)
ffn_output = self.fc1(out1)
ffn_output = self.act1(ffn_output)
ffn_output = self.fc2(ffn_output)
# ffn_output = self.ffn(out1)
ffn_output = self.dropout2(ffn_output)
return self.layernorm2(out1 + ffn_output)
尝试仅使用transformer、仅使用Bi LSTM和同时使用transformer和Bi LSTM后,结果发现同时使用transformer和bi LSTM,且bi LSTM层数为3时,预测效果最好。
3. 模型结果分析
本方案主要尝试了三个改进方向:
词向量嵌入:Bert预训练、 Embedding(paddle.fluid)、 one_hot;
序列特征学习:序列卷积(残差结构);
类机器翻译(计算每个位置的ACGU是点的概率,由于类似机器翻译,故称之为类机器翻译):LSTM、Transformer。
基于以上三个改进方向,通过组合模型,得到三种模型在A榜数据的表现:
Model1 |
Model2 |
Model3 |
|
RMSD on B board |
0.246 |
0.243 |
0.241 |
* 交叉验证后,model3的最好成绩为0.239
4. 总结分析
使用Bert作预训练没有起到太好的效果,未来需要在这方面做更深入的研究;此外,为了发挥模型的科研价值,需使用更大的数据集更深层地研究其化学含义及解释;再次,从这次比赛中也收获了很多经验, 今后将考虑进一步用深度学习和机器学习方法预测DNA、RNA和各种分子及材料的化学性质。
GitHub源代码:
https://github.com/ziyicui2022/FAAR-ML.git
季军团队方案详解
1. 方案介绍
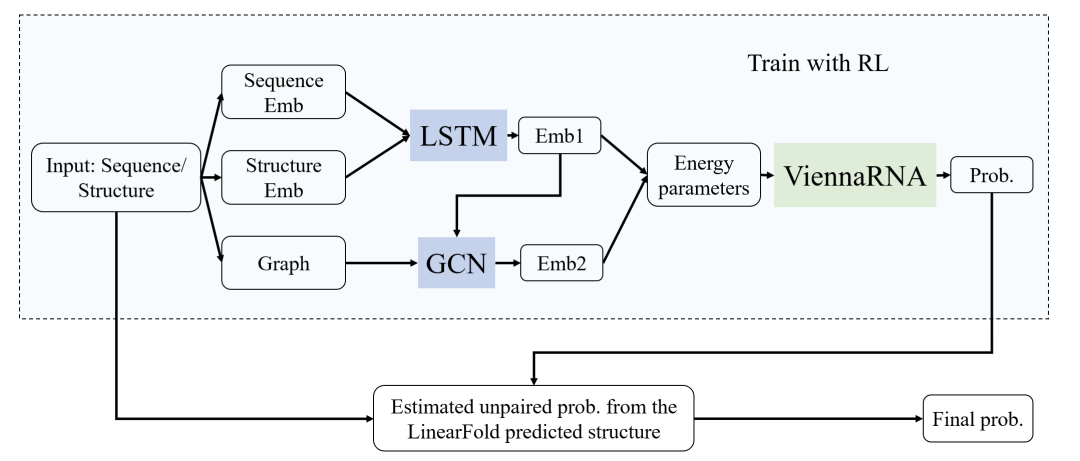
整体方案如下图所示,考虑到数据中同时有序列信息和图信息,分别使用双向LSTM提取序列信息,用图卷积神经网络GCN提取拓扑结构信息。由于训练数据较少,为降低过拟合的影响,先结合经典热力学方法和深度学习方法预测经典动力学方法中的能量参数(stacking energy),再使用ViennaRNA中提供的动态规划过程推测在给定能量下序列的不配对概率。
2. 模型搭建
首先,在飞桨中实现graphsage[2]。Graphsage包括聚合(aggregation)和更新两个阶段,聚合阶段使用平均池化,即:
其中
class egsage(Layer):
def __init__(self, in_channels, out_channels, edge_channels):
super(egsage, self).__init__()
self.in_channels=in_channels
self.out_channels=out_channels
self.edge_channels=edge_channels
self.message_lin=partial(paddle.fluid.layers.fc, size=out_channels, act="relu")
self.agg_lin=partial(paddle.fluid.layers.fc, size=out_channels, act="relu")
def forward(self, x, edge_attr, edge_index):
m_j=fluid.layers.gather(x, edge_index[1])
m_j=fluid.layers.reshape(paddle.fluid.layers.concat(input=[m_j, edge_attr], axis=-1), [-1, 1024+1])
m_j=self.message_lin(m_j)
#aggr
aggr_out=fluid.layers.scatter(x*0.0, edge_index[0], m_j)
aggr_size=fluid.layers.scatter(x*0.0, edge_index[0], m_j*0.0+1.0)
aggr_out=aggr_out/(aggr_size+1e-9)
#update
aggr_out=fluid.layers.concat(input=[x, aggr_out], axis=1)
aggr_out=self.agg_lin(aggr_out)
return aggr_out
然后,构建完整的神经网络模型。先将序列碱基和LinearFold预测结构中的点与括号嵌入为向量,再传入双向LSTM和GCN中提取信息,代码如下所示。
class Model(Layer):
def __init__(self, lr):
super(Model, self).__init__()
#Embedding
self.seq_embedding = partial(paddle.fluid.embedding, size=(4, 128), is_sparse=False)
self.bra_embedding=partial(paddle.fluid.embedding, size=(3, 128), is_sparse=False)
#Bidirectional LSTM
self.net1=partial(paddle.fluid.layers.dynamic_lstm, size=128*16, use_peepholes=False)
self.net2=partial(paddle.fluid.layers.dynamic_lstm, size=128*16, use_peepholes=False, is_reverse=True)
#GCN
self.gcn1=egsage(256*4, 256*4, 1)
self.gcn2=egsage(256*4, 256*4, 1)
self.gcn3=egsage(256*4, 256*4, 1)
#FCs
self.inp_fc=partial(paddle.fluid.layers.fc, size=128, act="relu")
self.seq_fc=partial(paddle.fluid.layers.fc, size=128, act="relu")
self.fc=partial(paddle.fluid.layers.fc, size=128*16, act="relu")
def forward(self, seq, dot, edge, edge_attr, fgt):
# Embed
emb_seq=self.seq_embedding(seq)
emb_dot=(self.bra_embedding(dot))
emb_dot=self.inp_fc(emb_dot)
emb_seq=self.seq_fc(emb_seq)
emb=self.fc(paddle.fluid.layers.concat(input=[emb_seq, emb_dot], axis=1))
# Bidirectional LSTM
emb1, _=self.net1(emb)
emb2, _=self.net2(emb)
emb=paddle.fluid.layers.concat(input=[emb1, emb2], axis=1)
# GraphSage
emb_g=self.gcn1(emb, edge_attr, edge)
emb_g = self.gcn2(emb_g, edge_attr, edge)
emb_g = self.gcn3(emb_g, edge_attr, edge)
# Final embedding
emb=paddle.fluid.layers.concat([emb, emb_g], -1)
3. 模型优化
由于A榜和B榜测试集分布差异较大,我们在A榜和B榜采用不同的模型优化策略。A榜数据集,由于RNA长度较长,动态规划耗时较长,可直接使用神经网络预测不配对概率,损失函数为预测值和基准值之间的RMSD。B榜数据集,可结合经典的动力学方法和深度学习方法,对RNA序列得到神经网络预测的一组能量参数
训练优化目标为降低的碱基不配对概率预测值和基准值之间的RMSD,主要难点在于动态规划过程不可导,不能直接使用梯度回传。为解决参数优化问题,参考强化学习训练技巧,对每个能量参数
其中
4. 结果分析
优化后模型的预测结果表明,对LinearFold预测结构中配对的位点,本方案预测的不配对概率绝大多数位于0附近(左图);反之,对LinearFold预测结构中不配对的位点,本方案预测的不配对概率绝大多数位于1附近(右图),与直觉一致。
在比赛过程中,少数较长的、预测结果与LinearFold预测的配对情况相差较大的序列对最终成绩的影响较大,如第13条RNA序列和第20条RNA序列。为进一步提高分数,我们将LinearFold给出的结构转化为二值的不配对概率,然后比较模型预测的不配对概率P与
最终在结合了双向LSTM、GCN、强化学习训练技巧和对预测值的提炼后,本方案在A榜阶段获第一名,B榜阶段获第三名。
GitHub源代码:
https://github.com/Zoesgithub/Paddlepaddle-RNA-structure-prediction-3rd-place-.git
往期回顾:
参考文献
如在使用过程中有问题,可加入官方QQ群进行交流:793866180。
如果您想详细了解更多飞桨的相关内容,请参阅以下文档。
·飞桨官网地址·
https://www.paddlepaddle.org.cn/
·飞桨开源框架项目地址·
GitHub: https://github.com/PaddlePaddle/Paddle
Gitee: https://gitee.com/paddlepaddle/Paddle
长按上方二维码立即star!
飞桨(PaddlePaddle)以百度多年的深度学习技术研究和业务应用为基础,集深度学习核心训练和推理框架、基础模型库、端到端开发套件和丰富的工具组件于一体,是中国首个自主研发、功能丰富、开源开放的产业级深度学习平台。飞桨企业版针对企业级需求增强了相应特性,包含零门槛AI开发平台EasyDL和全功能AI开发平台BML。EasyDL主要面向中小企业,提供零门槛、预置丰富网络和模型、便捷高效的开发平台;BML是为大型企业提供的功能全面、可灵活定制和被深度集成的开发平台。
END